Due to a huge influx of spam across all OBF wikis, we are in the process of locking down new user account creation and adding OpenID logins for the OBF wikis (BioPerl example). User account creation via the old login system will be disabled and OpenID will be the default path for new accounts so users to make wiki changes. This currently appears to have cut the incidence of spam significantly. We will be adding information to the login pages to redirect new users to the new login page.
[Read More]OBF Redmine server now available
The OBF now has a sparkly new Redmine instance running on Amazon EC2, thanks to efforts from Chris Dagdigian and Jason Stajich (with some admin help from yours truly). Bugs and user names (along with email contacts) from our old Bugzilla v2 server have been migrated over, though some links need to be fixed.
Redmine is a project management web application that has several nice features over other systems, including issue tracking, multiple project management, wikis, forums, and calendaring.
[Read More]BioPerl has moved to GitHub
BioPerl has migrated to git and GitHub! We have also set up a mirror set of several key repositories at the great public git hosting site repo.or.cz.
If you are a current BioPerl developer (had a previous account for direct access to our prior Subversion repository), please sign up for a GitHub account and let us know your user ID. Also, add the extra email 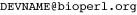 (where ‘DEVNAME’ is your original Subversion account ID). This should map any previous commits from the older Subversion and CVS repository to your new GitHub account.
(where ‘DEVNAME’ is your original Subversion account ID). This should map any previous commits from the older Subversion and CVS repository to your new GitHub account.
O|B|F Google Summer of Code Accepted Students
I’m pleased to announce the acceptance of OBF’s 2010 Google Summer of Code students, listed in alphabetical order with their project titles and primary mentors:
Mark Chapman (PM Andreas Prlic) - Improvements to BioJava including Implementation of Multiple Sequence Alignment Algorithms
Jianjiong Gao (PM Peter Rose) - BioJava Packages for Identification, Classification, and Visualization of Posttranslational Modification of Proteins
Kazuhiro Hayashi (PM Naohisa Goto) - Ruby 1.9.2 support of BioRuby
Sara Rayburn (PM Christian Zmasek) - Implementing Speciation & Duplication Inference Algorithm for Binary and Non-binary Species Tree
[Read More]O|B|F in Google Summer of Code
The Open Bioinformatics Foundation has been accepted as a mentoring organization for this summer’s Google Summer of Code. Our list of project ideas and mentors is linked from the O|B|F GSoC page.
Student applications must be submitted to Google by April 9, 2010, see the official GSoC 2010 FAQ. That is less than three weeks away!
[Read More]BioPerl at GMOD Meeting 2010
BioPerl developers and users attended the BioPerl satellite meeting on January 13th, just prior to the GMOD Meeting. Several items were covered on the agenda:
- In order to start addressing whole genome data with more lightweight objects, we are planning on setting up a lightweight Bio::SeqI object that has a flexible DB backend (i.e. Bio::DB::SeqFeature::Store or similar). We are also contemplating adding lazy parsing for some parsers, possibly using the Bio::PullParserI methods (or similar) that Sendu Bala created.
- After a final 1.6 branch point release, we may ‘freeze’ BioPerl in a maintenance mode, primarily so that we can reorganize core into several more easily installed subdistributions on a branch. New modules will essentially be additional separate repos that will depend on BioPerl core. This reorganization has been discussed for a few years now, and as we edge closer to starting this (probably this spring) we’ll announce more details.
- Some initial thoughts on how to handle circular genomes more efficiently. We essentially do this already, but it isn’t full-proof.
- Need some significant time dedicated towards GFF3-based coding (reimplement FeatureIO but allow some flexibility). Rob Buels had started the initial run at splitting out FeatureIO, so next step is a true reimplementation.
- We don’t plan on including Moose support for the immediate future, feeling that it would be better to reimplement some of the classes from scratch using Moose and similar as a BioPerl 2.0, or possibly await the impending Rakudo Perl 6 alpha and start afresh using that instead of Moose.
Anything we missed? Anything you would like to address? Please add comments and we’ll discuss them on list.
[Read More]Sanger FASTQ format and the Solexa/Illumina variants
I’m delighted to announce an open access publication in Nucleic Acids Research describing the FASTQ file format based on the conventions agreed by the OBF projects:
The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants Peter J. A. Cock ( Biopython), Christopher J. Fields ( BioPerl), Naohisa Goto ( BioRuby), Michael L. Heuer ( BioJava) and Peter M. Rice ( EMBOSS). Nucleic Acids Research, doi:10.1093/nar/gkp1137
This will hopefully serve as a reference describing the original standard Sanger FASTQ, and the two variants from Solexa/Illumina, and how to inter-convert between them.
[Read More]BioPerl interview in latest FLOSS Weekly
Two of the core BioPerl developers, Jason Stajich and Chris Fields, were interviewed by FLOSS Weekly. The interview is now available as an MP3 on the FLOSS Weekly website; several streaming versions (including podcast) are also available.
BioPerl core 1.6.1 PPM available
BioPerl 1.6.1 is now available for ActivePerl as a PPM, instructions for downloading can be found on the BioPerl wiki. This has been tested only for ActivePerl 5.10 and above, so any feedback with older versions of BioPerl would be greatly appreciated.
First 1.6.1 alphas of BioPerl-Run, BioPerl-DB, BioPerl-Network
Running a bit late on this, so just a quick note that the first alphas for BioPerl-Run, BioPerl-DB, and BioPerl-Network have been uploaded to CPAN:
They can also be downloaded from the BioPerl website:
This is the first run where we’ve switched to a regular Module::Build installation, so expect some initial bumps! There are a few initial problems that I plan on addressing soon, the main one being none of the modules are assigned version numbers (this may be a consequence of not pulling the version from a specific module). The other, more serious one, is that the Build.PL script checks for DBI but isn’t checking for any compatible DBD::* adaptors for BioPerl-DB (so it fails tests if DBI is installed). We have code in core to check for DBI drivers, so I may adapt that for BioPerl-DB.
[Read More]