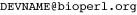The Biopython team is proud to announce Biopython 1.54, a new stable release of the Biopython library. Biopython 1.54 comes five months after our last release and brings new features, tweaks to some established functions and the usual collection of bug fixes.
This is the first stable release to feature the new Bio.Phylo module which can be used to read, write and take data from phylogenetic trees in Newick, Nexus and PhyloXML formats. The module is the result of Eric Talevich’s Google Summer of Code project which was supported by The National Evolutionary Synthesis Center (NESCent).
[Read More]